

Abstract
Introduction: Hypereosinophilic syndrome is characterized by sustained blood and/or tissue hypereosinophilia leading to tissue damage and organ dysfunction. Nearly half the cases are classified as idiopathic, since no known cause is identified and there is no evidence of a clonal disorder. In such cases, nonspecific treatment approaches, including corticosteroids, hydroxyurea and interferon-alpha, are used aimed at mitigating eosinophil-mediated organ damage. However, mortality occurs primarily due to cardiac and thromboembolic complications. Understanding the cause and mechanism of disease would aid in the development of targeted therapies with greater efficacy and fewer side effects. For instance, while the traditional view is that eosinophils are detrimental via toxicity of their granule proteins, recent studies suggest other mechanisms including non-canonical cell death subroutines, such as regulated necrosis, release of pro-thrombotic DNA "traps" and others. Lack of a mouse model hinders testing such hypotheses in vivo. We have developed a mouse model of eosinophil-associated hypereosinophilic syndrome characterized by multi-organ involvement including lethal prothrombotic myocardial damage.
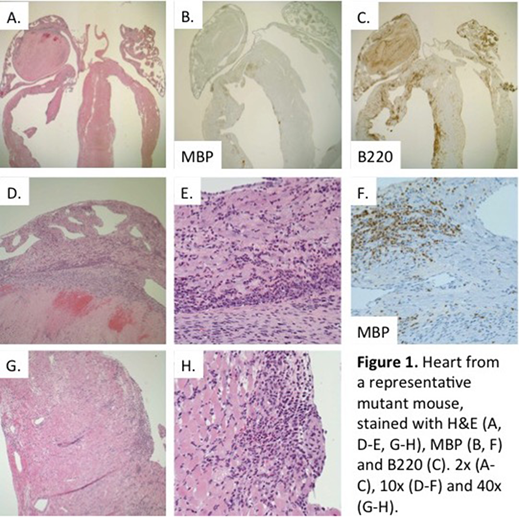
Results: We discovered a spontaneous mouse mutant in our colony with a hypereosinophilic phenotype. Mice develop peripheral blood eosinophilia, infiltration of lungs, spleen and heart by eosinophils, and extensive myocardial damage and remodeling. This ultimately leads to heart failure and premature death, often by 14 weeks of age. Histopathologic assessment of the hearts from affected mice (Fig. 1) revealed a robust inflammatory infiltrate of the ventricular myocardium. Immunohistochemistry demonstrated that this infiltrate is composed primarily of eosinophils (MBP) and B lymphocytes (B220), leading to myocardial damage and replacement fibrosis. In some cases, hearts showed dilatation and thinning of the right ventricular wall, suggestive of an inflammatory dilated cardiomyopathy. An exam of most mice revealed primarily atrial thrombi, which often filled the entire/majority of the chamber. Expression analysis by protein multiplex assay in heart homogenates revealed overexpression of Th1 and Th2 cytokines and chemokines, including IL-4, IL-6, LIF, IL-16, IP-10/Cxcl10, MIG/Cxcl9, Eotaxin-1/Ccl11, RANTES/Ccl5, MIP-1β/Ccl4, MIP-3β/Ccl19, MCP-1/Ccl2, MCP-3/Ccl7, MDC/Ccl22 and TARC/Ccl17.
Conclusion: The pathologies observed in the mutant line are reminiscent of those seen in patients with hypereosinophilia, where cardiac-related morbidities, like congestive heart failure and thrombi, are the most common causes of death. As such, our model provides an opportunity to test mechanistic hypotheses and develop targeted therapies.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal